

Abstract
Introduction:
Fanconi Anemia (FA) is an autosomal recessive disorder characterized by bone marrow failure (BMF), constitutional anomalies and high risk of developing cancer. Distinguishing FA from severe aplastic anemia (SAA) can be challenging especially in asyndromic patients. We undertook a clinical and laboratory cohort study of adolescent and young adult (AYA) patients with a diagnosis of FA treated at our institution to characterize the clinical features in our population, and conducted a prospective translational study to explore integration of a genomic and proteomic approach for improved diagnosis and molecular characterization of FA.
Methods:
Data on FA patients was obtained from an institutionally approved BMF database and hematopoietic stem cell transplant (HSCT) database. Further data was obtained from a register of chromosomal breakage (CB) analysis results. Index cases were identified if they were older than 14 years of age at the time of diagnosis or under the care of adult hematology with a clinical diagnosis of FA based on the presence of BMF and abnormal CB, or clinical phenotype with the presence of homozygous FA related genes. Family pedigrees were constructed based on history.
In addition, patients presenting with BMF were enrolled onto an institutionally approved study investigating proteomic biomarkers and genomics of BMF syndromes. Consented peripheral blood samples and/or extracted DNA were subject to either panel based next generation sequencing (NGS) testing as part of the Saudi Genome Project or subjected to whole exome sequencing (WES) by external lab.
For proteomic analysis, peripheral blood plasma (PBP) samples from 6 patients with FA, 10 SAA patients and 7 normal controls were subjected to expression proteomics using liquid chromatography tandem mass spectrometry (LC-MS/MS).
Result:
Patients and clinical features: 55 patients (26 M, 29 F) in 30 families were identified. While 18 patients (32%) were referred with a diagnosis/suspicion of FA, in 26 (47%) FA was diagnosed at our institution. The most frequent anomaly was short stature (14 patients, 25%), skin changes (7, 12%), urogenital abnormalities (7, 12%), dysmorphism/craniofacial abnormalities (7, 12%), hands anomalies (4, 7%); 12 (22%) had no recorded anomalies. 18 patients (33%) developed a malignancy either before or after diagnosis of FA: solid tumors in 5 (9%), AML and/or MDS in 15 (27%); 3 (5%) of these patients had both solid tumors and AML/MDS.
Diagnostic Tests: 35 patients (63.6%) had a positive CB analysis with diepoxybutane (DEB) or mitomycin-C (MMC) testing; in 5 patients (9%) DEB testing was borderline and 3 (5%) had a normal CBA but had a diagnostic phenotype+/- family history and presence of a homozygous mutation in a known FA related gene. 14 patients had cytogenetic abnormalities and abnormalities involving chromosome 1 were the most frequent (50%).
Mutation Analysis: Mutational analysis was available for 12 (22%) cases; homozygous mutations in FA genes were identified in 10 patients (18%) in 7 families (23% of families): FANCA (5 patients/3 families); BRIP1 (2/2); FANCP (1/1); FANCD2 (2/1). In one case, post matched sibling (HSCT) blood sample revealed a known pathogenic heterozygous c.2632G>C,p.Glu878Gln mutation in FANCA, suggesting a carrier donor.
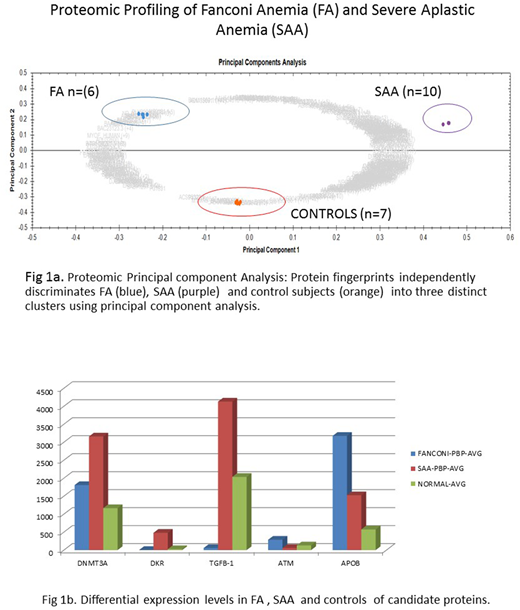
Proteomic analysis: Over 1650 unique PBP protein species were identified of which 605 were significantly differentially expressed (≥ 2 to ∞ - fold change & p < 0.001) between SAA /FA/ normal control subjects (Fig 1a). DNMT3A, Kinase Insert Domain Receptor (KDR) and TGFB-1 was found to be highly expressed in SAA versus FA, while ATM and APOB were highly expressed in FA versus SAA (Fig.1b).
Treatment outcomes: 36 out of 55 patients (65%) received HSCT. Actuarial survival of HSCT (n=37) and non-HSCT (n=14) patients was 70% and 77%, respectively. Treatment details were not available on 6 (11%).
CONCLUSION:
We report the first characterization of AYA patients with FA in Saudi Arabia. Our report emphasizes the need for a high index of suspicion of a diagnosis of FA in BMFs. CB may be falsely negative in cases, and panel based and/or WES based NGS testing increases diagnostic accuracy; in this cohort, mutations in FANCA were the most frequent (50%). Occurrence of hematological and solid tumors is a significant risk in these AYA patients. We also report proteomic panels as potential biomarkers that distinguish FA from SAA and may provide mechanistic insights.
No relevant conflicts of interest to declare.

